A.S.H
 |
Seung-Pyo Lee Seoul National Univ. Hosp. |
Overview of Amyloidosis >> |
1. Definition and Prevalence
Cardiac amyloidosis is caused by abnormal deposits of non-native, insoluble proteins in the heart mainly in the interstitial space of the myocardium but also in the vessels, valves and the pericardium. Approximately 10 proteins are known to cause cardiac amyloidosis with light chain immunoglobulin, wild type transthyretin (TTR) or mutant TTR being the most common. An amyloid protein is noted with a letter ‘A’ in front of the amyloid protein of interest, for example, an amyloid form of TTR as ATTR and the amyloid light chain as AL.Cardiac amyloidosis is rare but because of the unique morphologic and also, the possibility of cure, is conceived as treatable disease. The prevalence of AL type is approximately 1/10 million in the US and is almost undoubtedly associated with plasma cell dyscrasia. The prevalence of ATTR type has been reported to be up to 25% in those with 80 years or older by autopsy and certain reports have demonstrated that wild type TTR (wtTTR) cardiac amyloidosis might be underestimated in the elderly population. Considering these findings, the prevalence of cardiac amyloidosis may be underestimated than expected.
2. Diagnosis of Cardiac Amyloidosis (Table 1)
The gold standard method of diagnosing cardiac amyloidosis is to verify the presence of amyloidogenic proteins in the heart that may explain the organ dysfunction. However, the diagnosis of cardiac amyloidosis can be made without endomyocardial biopsy if the amyloid is verified in an organ other than the heart and if the ventricular thickening or the rise of serum N-terminal pro-B-type natriuretic peptide (NT-proBNP) cannot be explained otherwise.But the use of the invasive tools for definite diagnosis, it is more important not to miss certain clues that leads to the clinical suspicion of cardiac amyloidosis. Cardiac amyloidosis is often diagnosed late because of vague symptoms that range from exertional dyspnea, (pre)syncope, chest pain or even sudden cardiac arrest, which may sometimes seem unclear and vague, leading to significant delays in the appropriate diagnosis and even, treated inappropriately for the wrong disease. The following are some typical findings that may be encountered in noninvasive tests and give clues to the diagnosis of cardiac amyloidosis. Importantly, the clinical presentation of cardiac amyloidosis is very diverse as stated previously and therefore, the absence of the listed findings does not rule-out its absence.
2.1. Electrocardiogram
There may be any abnormal 12-lead electrocardiogram (ECG) finding in up to 90% of patients with cardiac amyloidosis. A typical finding is the low QRS voltage in the limb leads despite ventricular thickening (Figure 1), which is extremely rare in patients with other disease that present as ventricular hypertrophy. A QS wave in any two consecutive leads, especially in the septal leads, is called ‘pseudoinfarction’ as it is not a real ‘infarction’. The pseudoinfarction pattern is also a 12-lead ECG finding that can be commonly seen in cardiac amyloidosis patients (Figure 2). However, the prevalence of the typical electrocardiographic findings is <50% and therefore, the low QRS voltage nor the pseudoinfarction pattern may not be seen in a majority of cardiac amyloidosis cases.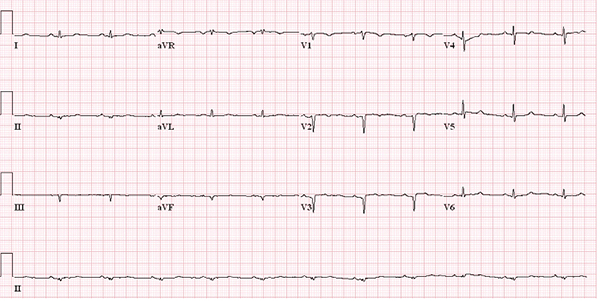
Figure 1. A typical low QRS voltage finding in a cardiac amyloidosis patient.

Figure 2. A typical pseudoinfarction pattern in lead V1 and V2.
2.2. Echocardiography
Cardiac amyloidosis should be always suspected in patients with symptoms or signs of heart failure, especially when the ventricular wall is thick (Video 1) and when there is prominent diastolic dysfunction (Video 2). Cardiac amyloidosis may present as concentric hypertrophy in contrast to hypertrophic cardiomyopathy where localized ventricular thickening is more common. However, there have been papers that describe cardiac amyloidosis without ventricular thickening.Echocardiography is very useful for patients with cardiac amyloidosis because it serves as a gatekeeper to the diagnosis of cardiac amyloidosis in most institutions and it can evaluate not only the structure of the heart but also, the hemodynamic consequences associated with the disease. It is not uncommon to notice findings other than the ventricular thickening such as valvular thickening and pericardial effusion. Biatrial enlargement that reflects the chronic diastolic dysfunction is seen in a majority of the patients.
One of the most striking finding may be the ‘apical sparing’ pattern on the bull’s eye plot of the global longitudinal strain using speckle tracking echocardiography (Figure 3). Although in a single center study, this apical sparing pattern has a 80~90% sensitivity and specificity for the diagnosis of cardiac amyloidosis.
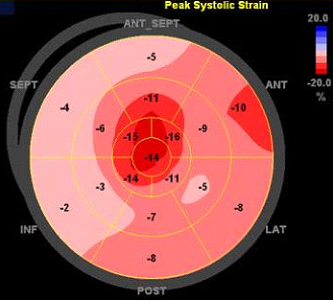
Figure 3. Apical sparing pattern on strain imaging that is typical for cardiac amyloidosis.
2.3. Cardiovascular magnetic resonance (CMR)
With the advent of new sequences and clinical experiences, CMR is now used popularly in patients with cardiac amyloidosis. It is useful not only for evaluation of the wall thickness and the overall structure associated with the disease but is unique for evaluation of the myocardial change itself, which cannot be fully evaluated with any other technology.Using gadolinium-based contrast, the discovery of the difference in the gadolinium kinetics in the myocardium between the normal versus the amyloidosis patients has been seminal. The pattern of late gadolinium enhancement (LGE) in the myocardium is may be diverse, ranging from the global transmural or patchy focal LGE to suboptimal myocardial nulling (Figure 4), which has been shown to match the deposition pattern of the interstitial amyloid. Incidental findings such as intracardiac thrombus may also be detected on CMR, because of its superiority in signal-to-noise ratio over that of the echocardiography.
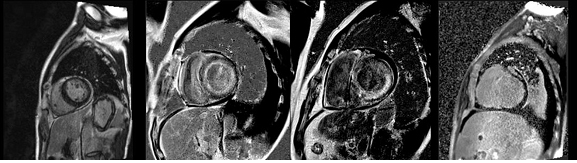
Figure 4. LGE-CMR findings, ranging from normal (far left), subendocardial diffuse LGE (middle left), patchy transmural LGE (middle right) and suboptimal myocardial nulling (far right).
The parametric CMR has also been investigated for the assessment of cardiac amyloidosis and is now used as adjunct methods in the evaluation of the disease. This is based on the histologic findings that the extracellular space is significantly expanded in cardiac amyloidosis than any other myocardial disease and that this finding is reflected by the lengthening of the myocardial T1 relaxation time. Using specific sequences for mapping the T1 relaxation time such as modified look-locker inversion recovery (MOLLI) sequence or shortened MOLLI (shMOLLI) sequence, the measurement of native T1 or the calculation of extracellular volume fraction from both pre- and post-contrast T1 mapping results may also help in the diagnosis of cardiac amyloidosis (Figure 5).
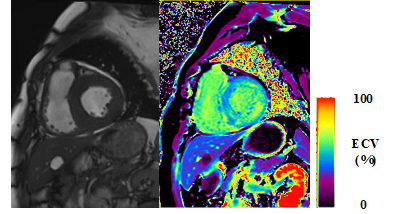
Figure 5. A patient with thick ventricle in cardiac amyloidosis (left panel) and a representative ECV map (right panel). The average ECV of the LV myocardium was approximately 50%
2.4. Nuclear imaging
2.4.1. Single photon emission computed tomography (SPECT)
Bone imaging SPECT tracers based on diphosphonates, such as 99mTc-pyrophosphate (99mTc-PYP), 99mTc-methylene diphosphonate (99mTc-MDP), 99mTc-hydroxymethylene diphosphonate (99mTc-HDP) and 99mTc-3,3-diphosphono-1,2-propanodicarboxylic acid (99mTc-DPD) can be used for the diagnosis of cardiac amyloidosis, especially the ATTR type (Figure 6). Of these, the 99mTc-DPD has been most extensively investigated and it does not seem to differentiate between the wtTTR versus the mtTTR. Although accurate quantitation of the tracer retention is difficult using SPECT, the degree of tracer retention in the myocardium is a marker of the myocardial involvement severity.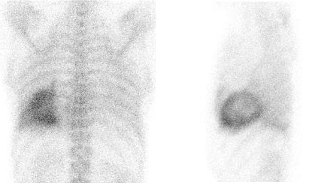
Figure 6. A representative 99mTc-DPD scan in a patient with wtTTR cardiac amyloidosis
2.4.2. Positron emission tomography (PET)
The recently developed PET tracers have been made to directly target the amyloidogenic proteins itself. Building on the fact that the cause of Alzheimer’s disease is amyloid accumulation in the brain parenchyme, several investigators have reported the clinical utility of amyloid PET tracers in cardiac amyloidosis such as 11C-Pittsburgh B (PiB) compound in imaging cardiac amyloidosis. Others have also used 18F-tagged radiotracers such as 18F-florbetapir or 18F-florbetaben because of the limited half-life of the 11C (t1/2≈20minutes) and the relatively longer half-life of the 18F (t1/2≈110minutes). These 18F-tagged radiotracers have been shown to accumulate specifically to the sites of amyloid deposits in the myocardium. The major caveat with the PET agents is that there is a shortage of outcome studies that show its clinical utility.2.5. Pathologic diagnosis
The visualization of amyloid deposit directly from pathologic specimens is the cornerstone of cardiac amyloidosis diagnosis. A typical myocardial specimen of the cardiac amyloidosis patient would show expansion of the extracellular matrix in a conventional hematoxylin and eosin staining (Figure 7). This amyloid deposit is confirmed by a typical apple-green birefringence on Congo red staining (Figure 7). However, some institutions advocate the use of adjunct methods such as sulfated Alcian blue or amyloid P immunostaining as the sensitivity may be low and the difficulty in quantification of amyloid deposit in the Congo red staining. The type of amyloid deposit can also be confirmed with immunohistochemical staining. The pattern of amyloid deposit may vary, from a typical diffuse subsarcolemmal pattern to ‘nodular’ patterns where amyloid deposits can be seen in the form of clumps in the interstitial space.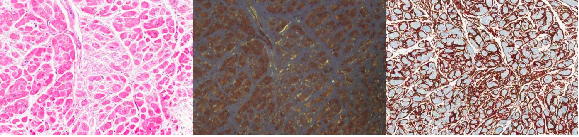
Figure 7. Myocardial specimen examination by pathology. H&E (left), Congo-red (middle), amyloid P (right) staining results.
2.6. Typing cardiac amyloidosis
It is important to determine the type of cardiac amyloidosis as this significantly impacts the treatment and also the prognosis. The typing of AL cardiac amyloidosis depends on the analysis of the serum protein, for example, protein immunoelectrophoresis and serum free light chain assay. However, there are some important caveats to consider with these ‘indirect’ methods. First, these ‘indirect’ methods of typing are only useful in systemic AL amyloidosis and furthermore, there may be some AL cardiac amyloidosis without a definite rise in the serum concentration of the AL protein. Second, although the majority of cardiac amyloidosis which are not the AL type by serum light chain assay may be the ATTR type, a minority of cardiac amyloidosis patients are neither the AL nor the ATTR type. Therefore, it would be too impetuous to directly determine the type of cardiac amyloidosis without using either the immunohistochemical or the proteomic analysis or even both.Pathologic examination are very useful for determining the type but in contrast to the expectation, the immunohistochemical staining results may not be always clear-cut and there can always be false-positive or -negative results. A lot of laboratories may support these pathology results with appropriate proteomic analysis, mainly mass spectrometry. Additionally, in those who are suspected to have cardiac amyloidosis due to mtTTR, the genotyping of the TTR gene is also a must-do item.
3. Treatment
3.1. Treatment of heart failure symptoms
Besides the treatment of amyloidosis infiltration itself, the mainstay of cardiac amyloidosis treatment is directed towards control of heart failure symptoms. Loop diuretics, either stand-alone or in combination with mineralocorticoid antagonist, is often used for volume control. However, because a significant proportion of those with cardiac amyloidosis have concomitant autonomic dysfunction, diuretics should be used with caution so as to avoid excessive volume depletion. Likewise, based on the same reason, medications that are standard for heart failure, such as renin-angiotensin system blocker or beta-blocker, should be used prudently. The clinical evidence on the use of cardiac resynchronization therapy has not been proven, partially because of the limited survival in cardiac amyloidosis patients.3.2. Treatment directed against the amyloid protein
As AL cardiac amyloidosis is almost always invariantly associated with plasma cell dyscrasias, treatment of AL cardiac amyloidosis is directed against clearance of the plasma cells as well as the prevention of target organ damage, including the myocardium. The standard treatment regimen consists of a combination of chemotherapeutic agents, such as alkylating agent (melphalan, cyclophosphamide) and/or proteasome inhibitor (bortezomib) or immunomodulatory agent (thalidomide, lenalidomide). After a significant reduction of the plasma cell burden, the chemotherapy is usually followed by autologous hematopoietic cell transplantation, if eligible. However, as the chemotherapeutic agents have minimal effect on eliminating the AL protein deposited in the end organ, antibodies that direct the misfolded proteins are being actively developed.As for the ATTR, treatment is directed towards disruption of ATTR, reduction of TTR transcription or stabilization of the TTR protein. Following the promising results of doxycycline, together with tauroursodeoxycholic acid, in reducing amyloid aggregation in mice by stabilizing the TTR protein, a phase II trial has also shown promising results in cardiac amyloidosis. Anti-sense oligonucleotides directed against the transcription of TTR protein has also shown to be safe in phase II trials and are now being escalated into phase III trials. Tafamidis and diflunisal binds to the thyroxine binding sites of the TTR, thereby inhibiting the dissociation of TTR into monomers. Again, both have shown promising results in those with ATTR cardiac amyloidosis and in the midst of phase III clinical trials.
Although the cardiac involvement by amyloid protein was once considered a contraindication of heart transplantation, this belief is changing in centers where multidisciplinary team approach is possible. Heart transplantation is currently considered an option for cardiac amyloidosis in many hospitals nowadays and the outcomes comparable to any other restrictive cardiomyopathies.
Tables.
Table I. Diagnostic criteria for cardiac amyloidosis.
| Modality/Method | Parameter | Diagnosis of organ involvement |
| 1. Endomyocardial biopsy | Proof of amyloid deposit in the heart on pathologic examination | |
| 2. Biopsy of an organ other than the heart | Proof of amyloid deposit in other organs (fat pad, kidney, etc.) on pathologic examination | |
| 3. Echocardiography | Mean wall thickness | >12mm with no other cause and identification of amyloid deposit in other tissue or organ |
| 4. NT-pro-BNP | >332ng/L in the absence of renal failure or atrial fibrillation | |
For diagnosis of cardiac amyloidosis, the following criteria suffices.
1 only; 2 + 3 or 4.