A.S.H
 |
Hyung-Kwan Kim Seoul National Univ. Hosp. |
Overview of Hypertrophic CMP >> |
Definition
Hypertrophic cardiomyopathy (HCM) is a common genetic cardiac disease with an estimated prevalence of 0.2% (1:500) in the general population. HCM is morphologically defined by the presence of increased left ventricular (LV) wall thickness in the absence of hemodynamic causes explaining the degree of increased LV wall thickness such as hypertension or aortic stenosis. Adults with increased LV wall thickness resulting from cardiac amyloidosis or glycogen storage disease need to be excluded from the definition on the basis of clinical history, physical examinations, laboratory, imaging, and/or genetic tests. Histopathological feature of HCM includes myocyte disarray, however, this is not specific to autosomal dominant HCM and can be reposted in other forms of LV hypertrophy like Noonan’s syndrome and Freidreich’s ataxia, and even at the septal junction of LV and right ventricle of normal heart. Moreover, identification of myocyte disarray in vivo is challenging, and cannot be practically used in the daily practice. Genetic definition has also some limitations like issues of many different kinds of mutation sites and genotype-phenotype correlations. Therefore, for the clinicians, HCM is still defined on a morphological basis.Genetic basis
Genetic studies showed that HCM is caused by dominant mutations in genes encoding thick (actin) and thin (myosin) myofilament protein components of the sarcomere. Pathogenetic mutations causing HCM are transmitted in an autosomal dominant pattern. Mutations reported include β-myosin heavy chain (MYH7), myosin-binding protein C (MYBPC3), Troponin T (TNNT2), Troponin I (TNNI3), Troponin C (TNNC1), α-tropomyosin (TPM1) and so on.Clinical presentation
1) Sudden deathVentricular arrhythmia like ventricular fibrillation is known to be responsible for more than 80% of sudden death in HCM. Ventricular tachycardia, supraventricular tachycardia, atrial flutter or fibrillation can precede ventricular fibrillation. HCM sudden cardiac risk calculator is available at the website, http://www.doc2do.com/hcm/webHCM.html, and implantable cardioverter defibrillator should be considered in HCM patients at high risk for sudden death.
2) Dyspnea
This is the most common symptom that is observed in more than 90% of HCM patients. An increase in LV filling pressure is a main pathophysiology resulting from LV diastolic dysfunction. At the beginning, patients experience dyspnea on exertion, but with progression of LV diastolic dysfunction, progressive heart failure symptoms like orthopnea or paroxysmal nocturnal dyspnea can develop. Heart failure can develop in 10% of young HCM.
3) Syncope or presyncope
This is a symptom caused by a decrease in stroke volume or arrhythmia.
4) Angina
Anginal symptom in HCM can develop even in patients without coronary artery disease. Subendocardial ischemia in LV hypertrophy can be a potential explanation.
Diagnostic tests
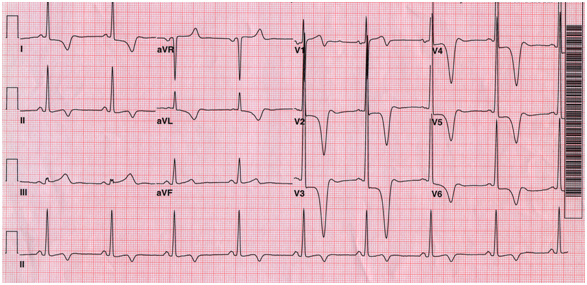
1) ElectrocardiogramST-T abnormality and LV hypertrophy are the most common findings on electrocardiogram. Of note, deep negative T wave inversion is typically shown in apical HCM (Figure 1).
Figure 1. Typical electrocardiogram in apical hypertrophic cardiomyopathy
2) Echocardiography
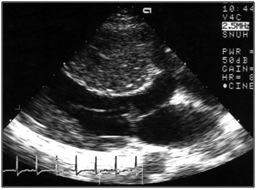
This is the most important diagnostic and prognostic imaging modality. Echocardiography can demonstrate abnormalities in anatomy, function, and hemodynamics of the heart. The typical echocardiographic findings can be summarized as follows (Figure 2); i) increased LV wall thickness and small LV, ii) LV systolic function is usually normal, but LV diastolic function is impaired, iii) decreased mitral annular early diastolic velocity, iv) LV outflow tract dynamic obstruction can be demonstrated by Doppler examination, v) systolic anterior motion of mitral valve and associated mitral regurgitation.
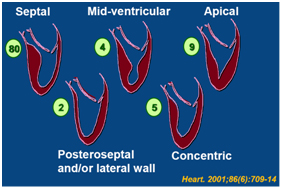
HCM can be classified according to the hypertrophic segments. The prevalence of apical HCM approaches 30% in the East Asian population
Figure 2. Typical echocardiographic findings in hypertrophic cardiomyopathy
| A. Two dimensional echocardiography | B. Systolic anterior motion of mitral valve |
 |
 |
C. Continuous wave Doppler shows dynamic obstruction in the LV outflow tract ; Dagger shape appearance.
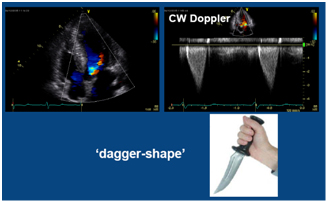
Figure 3. Classification of HCM (numerical values denotes prevalence)
3) Cardiac magnetic resonance (CMR)
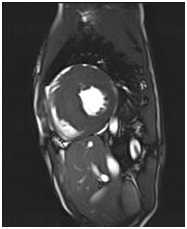
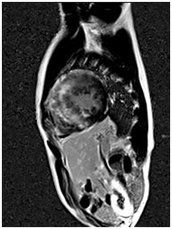
CMR is the best imaging modality to investigate myocardial fibrosis by using delayed enhancement technique (Figure 4). CMR-based assessment of myocardial fibrosis is expected to be used for predicting sudden cardiac death risk.
Figure 4. A 23-year-old HCM patient showing delayed enhancement on CMR.
| A. Septal hypertrophy | B. Fuzzy delayed enhancement at the hypertrophic sites |
 |
 |
Treatment
The prognosis is known to be genrally good, however a few patients at high risk for sudden death should be closely followed.1) Medical treatment
Beta blockers, Calcium-channel blockers, Disopyramide (not available in Korea), Amiodarone for arrhythmia
2) Intervention
Percutaneous alcohol septal ablation, Pacemaker insertion
3) Surgical treatment
Septal myectomy